Cystic fibrosis is a congenital metabolic disease. Body fluids such as saliva, bronchial mucus or pancreatic secretions are much tougher than usual due to genetic predisposition. Consequences include respiratory problems and indigestion. Cystic fibrosis is not curable. With consistent therapy, however, the course of the disease can be slowed down. Read here which symptoms cause cystic fibrosis and how to treat it.

Cystic fibrosis: short overview
- Description: hereditary metabolic disease, causing tough mucus formation in the lungs and other organs
- symptoms: Respiratory problems, irritating cough, lung infection, failure to thrive, indigestion, severe diarrhea, fatty liver, reduced fertility
- Causes: Inheritance of defective genes that affect the consistency of body fluids, outbreak of the disease only if both parents inherit a diseased gene (dominant recessive inheritance)
- Diagnosis: Blood test for immunoreactive trypsin (IRT), pancreatitis-associated protein (PAP), sweat test, genetic test
- Treatment: mucolytic agents, bronchodilating agents, inhalation, antibiotics for infections, cortisone, CFTR modulators, lung transplantation
- Forecast: not curable, course strongly dependent on the severity and timing of the diagnosis, shortened life expectancy
Cystic fibrosis: description
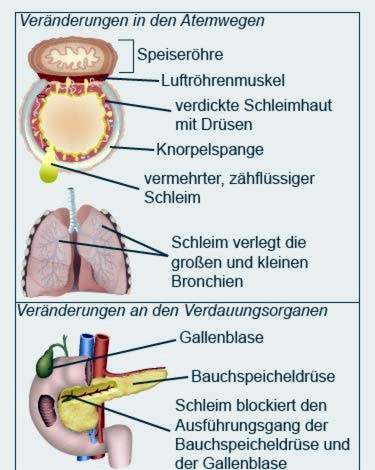
Cystic fibrosis (also called cystic fibrosis) is a hereditary metabolic disease. The formation of various body fluids is disturbed. The secretions of the lungs, pancreas and other organs are more viscous than in healthy people.
Tough mucus
The tough mucus clogs, among other things, the small branches of the bronchi and the ducts of the internal organs. Breathing and digestion are particularly affected. In the course of the disease the organs can work worse and worse.
Error in the genetic material
The cause of the disease are defects in the genetic material. Cystic fibrosis is therefore not curable. Time of diagnosis and severity of symptoms can vary widely individually. In many children cystic fibrosis makes a massive impact from birth, in other cases, it is recognized later.
Cystic fibrosis: symptoms
Cystic fibrosis symptoms can vary greatly from patient to patient. The disease affects the function of various organs, but especially the lungs and the digestive system.
Recognize early signs
The first symptoms of cystic fibrosis vary individually. In most cases, cystic fibrosis symptoms appear within the first year of life. Thus, the disease can usually be diagnosed early and be started quickly with a therapy. However, some patients have significant complaints only in adolescence. Not every affected person shows the full range of possible symptoms. The severity of the symptoms also varies.
Altered body fluids
In cystic fibrosis, the formation of the so-called chloride ion channels in the cells is disturbed. This changes the composition of body fluids. The easiest way to detect this change in the sweat of those affected. Your sweat is saltier than healthy people. The salts sodium and chloride, which belong to the so-called electrolytes, are enriched in their sweat. As a result of sweating, patients suffering from cystic fibrosis increasingly lose body salts.
Varied cystic fibrosis symptoms
The disease affects a whole range of organ systems. Often, the first symptoms of cystic fibrosis appear in the lungs and digestive tract. In the course of life further complaints can be added. Through targeted therapy, the symptoms can be treated well. However, the symptoms can also reach menacing proportions. It is especially dangerous when the bronchi clog through the viscous mucus. Then the patients can suffocate in extreme cases.
Cystic fibrosis: symptoms of the lung
Breathing problems and irritating cough
In most cases, in cystic fibrosis symptoms of the lungs occur only in slightly older infants. Newborns usually have no breathing problems. Cystic fibrosis symptoms are often expressed in the form of a cough-throat-like, chronic, irritating cough in slightly older children. The mucus in their respiratory tract is increased, tough and viscous. This obstructs the airflow in the lungs. Over time, a progressive respiratory distress develops.
Frequent infections
Increased mucus production in the lungs makes it easier for bacteria to colonize and cause infection. Recurrent pneumonia or bronchial infections are mainly caused by bacteria such as staphylococci and Pseudomonas species. The disturbed salt balance in the lungs also hinders the body’s defense. Also, pulmonary haemorrhages may occur. A typical sign of this is the coughing up of blood-mixed mucus.
Although the lungs are damaged from an early age, the first symptoms of cystic fibrosis in the respiratory tract are often only at primary school age or even later. The symptoms sometimes only occur when large parts of the lung tissue have already been destroyed or the respiratory tract is severely constricted.
Cystic fibrosis: symptoms of the pancreas
In patients with cystic fibrosis, the pancreas often becomes inflamed. It secretes a secretion that contains, among other things, enzymes for fat and sugar digestion. In cystic fibrosis sufferers, the secretion due to its viscosity condenses back and causes inflammation.
As the process progresses, the pancreatic tissue becomes hardened and scarred. Doctors speak of fibrosis. The fibrosis gradually destroys the pancreas. In addition to the bile, the pancreas also forms insulin, which is needed, among other things for the sugar utilization in the body. Patients who are a little older (from adolescence) often develop diabetes mellitus.
Cystic fibrosis: symptoms of bile
The pancreas and gallbladder share a common duct into the intestine. Therefore, the backflow of pancreatic secretions can also cause inflammation of the gallbladder. Often, gallstones form, which can completely block the gallbladder from the gallbladder.
Cystic fibrosis: symptoms of the digestive tract
In addition to complaints of the lungs, cystic fibrosis symptoms mainly affect digestion. Due to the lack of bile, for example, the fat digestion is impaired. Often, patients tolerate fatty foods poorly. The ingested food is largely excreted undigested again. Typical are then very voluminous and soft bowel movements.
Diarrhea and growth disorders
Affected children, including infants, often suffer from severe diarrhea. Although they drink and eat well, they barely increase. Growth disorders and malnutrition are therefore further classic consequences of the disease.
However, such complaints in the field of digestion can also occur in other diseases. Only in combination with respiratory problems are they therefore a characteristic indication of cystic fibrosis, which should definitely be investigated.
anal prolapse
In the further course, cystic fibrosis can cause various complications in the digestive tract. The most common is a so-called anal prolapse. In anal prolapse, the intestinal mucosa bulges out of the anus. Such an incident needs to be surgically treated as soon as possible.
bowel obstruction
Involvement of the intestine (invagination) or intestinal obstruction (ileus) is also common. Both complications are associated with severe abdominal pain and significant digestive problems. The pain usually occurs in spurts, especially after eating. An intestinal obstruction is lethal if left untreated. Spasmodic, acute abdominal pain should therefore always be clarified by a doctor.
Cystic fibrosis: symptoms of the liver
fatty liver
Under the backflow of bile also suffers the liver. In many patients, fatty liver develops during the course of the disease. Fatigue, loss of appetite, bloating and flatulence and, in rare cases, feelings of pressure or pain in the upper abdomen may occur.
cirrhosis of the liver
In rare cases, a shrinking liver develops (liver cirrhosis) in which the liver is severely disturbed in its function. It first manifests itself in the form of jaundice (jaundice). Signs of jaundice are the yellowish discoloration of white in the eyes. After a long time, heart problems also occur and the performance of those affected continues to decline.
Cystic fibrosis: reduced fertility
More than half of all male patients are infertile. Although they can form fertile sperm in most cases, they can not get through the vas deferens because they are blocked by viscous mucus.
Affected women are usually less fertile. In general, they can receive and deliver a child. However, in their fallopian tubes tough mucus accumulates, which the sperm can hardly penetrate. Especially at a higher age the probability of pregnancy decreases rapidly.
Cystic fibrosis: symptoms in children
Cystic fibrosis is a genetic disease. It has always been present since birth. But the classic cystic fibrosis symptoms do not always occur in childhood. However, there are often already nonspecific instructions that should be followed. This is especially true if cases of cystic fibrosis have already occurred in the family.
Bloated abdomen
An indication that there is a metabolic disorder is, for example, a bloated abdomen for a long time. Often the children suffer from diarrhea. In newborns, a markedly delayed first stool (Kindspech) may be an indication of cystic fibrosis. In many cases, growth and growth disorders occur even though the children eat with cravings. Only in rare cases it also leads to constipation (constipation) as a result of the disease.
Rattling breathing
Other symptoms that may indicate cystic fibrosis include rattling breathing and severe restlessness. Many children suffer from chronic inflammation of the sinuses. These are mainly noticeable by not exactly localizable pain in the face. Nasal polyps are more common in children with cystic fibrosis than in healthy children.
If children suffer from breathing problems or indigestion for longer, a doctor should always be consulted as a precautionary measure. In children, life-threatening situations can quickly occur because they themselves can not articulate their complaints or their severity can not assess.
Cystic fibrosis: causes and risk factors
Cystic fibrosis is caused by a genetic defect. The pathological change lies on the seventh chromosome in the so-called CFTR gene.
The CFTR gene (cystic fibrosis transmembrane regulator gene) contains the construction manual for a channel through which chloride ions enter the cells. The defective chloride ion channels block the transport of salt into certain body cells in cystic fibrosis patients.
The affected gland cells but instead of the otherwise liquid secretion tough mucus from. In the lungs, the paranasal sinuses, the pancreas, in the intestine, in the biliary ducts and in the gonads form so tough mucus secretions with high salinity.
Cystic fibrosis: how endangered is my child?
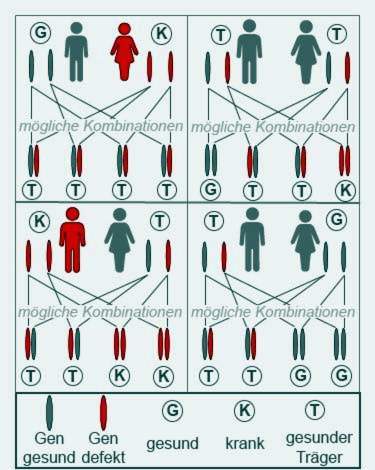
Cystic fibrosis only breaks out when both parents pass on a pathologically altered gene to their child. These parents are then mostly both themselves healthy, but carriers of the gene.
People with cystic fibrosis are only partially fertile. Some patients still become parents. Diseased fathers or mothers, however, always pass on a sick gene, as both CFTR genes carry the cystic fibrosis information. However, their children only get ill if they also get a sick gene from the other parent.
Couples whose families have already had cystic fibrosis cases should seek genetic counseling before planning a pregnancy.
Preimplantation genetic diagnosis
Parents who could give birth to cystic fibrosis can undergo preimplantation genetic diagnosis. In preimplantation genetic testing, the oocytes are first artificially fertilized. The first cell divisions take place in the test tube (in vitro).
Before inserting an embryo, it is tested for altered gene properties. Only embryos are then implanted that do not carry the diseased gene. Irrespective of this, it can also be examined during pregnancy whether the child will develop CF later.
Cystic fibrosis: examinations and diagnosis
Unlike a few years ago, most hospitals today routinely undergo neonatal screening. This includes studies on cystic fibrosis.
Screening in blood and sweat
For the screening, blood is taken from the newborn. The cystic fibrosis test involves several stages:
blood test
Test for elevated levels of immunoreactive trypsin (IRT) and pancreatitis-associated protein (PAP). In case of abnormalities, the sweat test takes place.
welding test
Cystic fibrosis patients have a significantly higher salinity in sweat than healthy people. For the cystic fibrosis sweat test, the content of the salts sodium and chloride in the body sweat is measured. In children, the sweat on the forearm is collected and then analyzed. If a suspicion arises here, a genetic test will be carried out.
Genetic test
In patients with cystic fibrosis, the so-called CFTR gene, which provides the blueprint for specific ion channels, has changed. This construction manual is long, it consists of about 6500 base pairs. Everywhere an error can creep into code, but the errors have different effects. Therefore, only the most common deviations in the code are tested.
Family history gives hints
If no appropriate neonatal screening has been carried out and the suspected cystic fibrosis comes up later, the family doctor or internist is the right person to contact. In an initial conversation, this person records the medical history (anamnesis). In case of suspected cystic fibrosis, particular attention is paid to the family history.
Physical examination
Subsequently, a physical examination takes place. The doctor listens to the lungs and scans the internal organs. He may already rule out some other conditions that are associated with symptoms similar to those of cystic fibrosis.
In addition, X-ray examinations may show respiratory and pulmonary blockages. Laboratory investigations give indications of functional restrictions of the internal organs. Even in adults who are suspected of cystic fibrosis, a sweat test provides important evidence for the diagnosis.
Testing family members
If cystic fibrosis is detected in a family, it makes sense that all other family members also undergo an examination. Cystic fibrosis can also occur in attenuated forms. It often takes many more years for cystic fibrosis to manifest itself with clear symptoms. Nevertheless, early diagnosis and cystic fibrosis therapy is also important for those affected to increase life expectancy.
Cystic fibrosis: treatment
Cystic fibrosis is not curable. Children born with cystic fibrosis suffer the effects of the disease throughout their lives. However, a combination of physiotherapy, medication and inhalations can significantly slow the progression of the disease. A cystic fibrosis therapy should therefore primarily achieve that those affected can lead as normal a life as possible.
Learning to live with the disease
Especially for children it is important to learn to deal with the disease in the long term. Children with cystic fibrosis should learn as early as possible what the disease means and how it affects the body. Here a hospital stay with special training units can be useful. In the process, children and parents learn how to feed themselves, what sport looks like, and how they behave best in critical situations.
Help for the lungs
There are various options for treating the symptoms. Depending on the age of the patient and the severity of the symptoms, different approaches are recommended.
Mucolytic agents
Cystic fibrosis patients suffer most from lung problems. By regular inhalation with special additives (mucolytics), the viscous mucus dissolves and can be easily coughed off.
Bronchial dilating agents
So-called beta-2-sympathomimetics also increase the bronchi, which additionally facilitates breathing.
Antibiotics against bacteria
Due to the poor ventilation of the lungs, people with cystic fibrosis suffer from bacterial infections of the respiratory tract much more frequently. On the other hand, antibiotics administered in good time help. In some cases, it makes sense to inhale these permanently.
Anti-inflammatory drugs
In many patients the respiratory tracts are often or chronically inflamed. Then anti-inflammatory drugs like cortisone help.
CFTR modulators
Meanwhile, the first drugs have been developed that improve the impaired function of the ion channels. However, they only work for certain mutations and thus only for a small part of the patients. Their effectiveness is also limited. The improvement of the drugs is intensively researched. For the first time, they would begin with the cause of the disease rather than just the symptoms.
Lung Transplant – the last hope
For severe cases, a lung transplant is also an option. As drastic as this step seems at first, many patients can lead a significantly lower-burdened life afterwards.
Feed properly in cystic fibrosis
Since cystic fibrosis also disturbs digestion, patients must pay close attention to their diet. You should prefer a diet high in protein and carbohydrates.
There are also vitamin supplements and minerals. The latter replace the salts that sweat the patients in large quantities. As the pancreas does not work properly, children are given digestive enzyme medications as an adjunct to meals.
Perceive vaccinations
Vaccination is particularly important for patients with CF. With them, bacteria have easier play and they often get more severe than patients who are not preloaded. Especially vaccines against measles and pneumococci are recommended. In addition, every year should be a flu vaccine.
Cystic fibrosis: disease course and prognosis
Cystic fibrosis is caused by a change in the genome and is therefore not curable. For people with cystic fibrosis, life expectancy and quality of life are usually significantly reduced. Without therapy worsens the state of health rapidly and those affected usually do not live long.
With timely and consistent therapy, the course of the disease can be significantly slowed down. In the meantime, patients live much longer than a few years ago. The mean life expectancy for cystic fibrosis is currently around 40 years. But many also live with the disease for 50 years and more.
Complications and sequelae
Even with intensive therapy, complications can occur again and again in cystic fibrosis. Most often there is acute respiratory distress due to poor lung ventilation. Individual areas of the lung can even collapse (alektase).
Frequently, chronic bronchitis or pneumonia develops. Mushrooms can also affect the lungs.
In addition, shifts in the fluid and electrolyte balance can trigger shock and circulatory failure.
Other complications and sequelae include:
- chronic liver disease, especially cirrhosis
- Gallbladder inflammations and gallstones
- chronic inflammation of the pancreas
- disturbed heart function
- acute intestinal obstruction (ileus)
- Intestinal invagination
- malnutrition
- Diabetes mellitus
- Limited fertility of women, or infertility of male patients
Cystic fibrosis is a hereditary disease, prevention is therefore not possible. People at risk of familial risk should seek genetic counseling if they wish to have children. A genetic test is performed and it is examined whether the CFTR gene is altered. Depending on whether one or both partners carry the gene, the risk for the offspring can be calculated.
In the meantime, in cystic fibrosis preimplantation genetic diagnosis (PID) is also possible in Germany. The prerequisite for this is always the approval of an ethics committee. Oocytes are fertilized outside the womb and only embryos without the problematic cystic fibrosis genes are used.
Additional information
guidelines:
- S2 Consensus Guideline “Diagnosis of Cystic Fibrosis” of the Society for Pediatric Pulmonology (2013)
- S3 Guideline “Pulmonary Disease in Cystic Fibrosis” by the Society for Pediatric Pulmonology (2013)
Support Groups:
- Cystic Fibrosis e.V.